
|
|
|
Главная -> Свойства координационных соединений ЛКАО) электроны. Эта часть энергии, существенная при абсолютных оценках (особенно при построении поверхностей потенциальной энергии, см. след. раздел), часто игнорируется при исследовании относительной устойчивости, так как принимается, что энергия остовов слабо зависит от изменения валентного окружения. Рассмотрим конкретный случай октаэдрического комплекса. На рис. V. 4 приведена примерная схема расположения МО (с учетом только а-связей), по которым необходимо распределить 12-\-п электронов. Первые 12 электронов располагаются на первых щести связывающих орбиталях aig, tiu и eg. Вносимый этими электронами вклад в устойчивость комплекса является основным. С ростом заряда ядра металла энергия на связывающих МО будет понижаться (ср. с ЭИВС, стр. 154). Отсюда следует, что основной вклад в устойчивость комплексов должен монотонно увеличиваться при следовании от одного переходного металла к другому по таблице Д. И, Менделеева слева направо. Дополнительный вклад в устойчивость могут внести оставщие-ся п электронов. Они должны распределяться по орбиталями gg из которых последняя - разрыхляющая. Эта часть устойчивости изменяется не монотонно: с увеличением количества электронов на eg-орбиталях, как уже отмечалось, она должна убывать, а не расти. При малом п, когда заполняются только 2е-орбитали, устойчивость растет *, но при достаточно большом п ( > 3 для высокоспиновых комплексов и > 6 для низкоспиновых) начинают заполняться разрыхляющие eg-орбитали и устойчивость убывает. При максимальном заполнении eg-орбиталей {п - Ъ для низкоспиновых комплексов и п = 10 для высокоспиновых) устойчивость комплекса минимальна. Этим можно объяснить сравнительно низкую устойчивость высокоспиновых комплексов Мп(11), Fe(IIl) и Zn(Il). Еще более наглядной становится картина относительных устой-чивостей комплексов ряда переходных металлов в терминах кристаллического поля. Основной вклад в устойчивость определяется здесь притяжением лигандов к сферически-симметричному остову центрального иона. Очевидно, что эта часть устойчивости должна расти монотонно (почти линейно) с ростом заряда. Иную картину представляют валентные электроны, электростатическое взаимодействие которых с лигандами существенно зависит от распределения электронного облака относительно лигандов. Как было указано в разделе IV. 5, с учетом расщепления термов в октаэдрическом поле появление электрона на 2е-орбитали связано с сообщением комплексу дополнительной устойчивости** * - орбита.пи в октаэдрических комплексах могут образовать связывающие я-орбитали (стр. 120) или оставаться несвязывающими. И в том и в другом случае они вносят дополнительную устойчивость в систему (в последнем случае - в виде электростатической экстрастабилизации, см. ниже). ** В силу сказанного ранее (раздел IV. 2) эту дополнительную устойчивость надо понимать в относительном смысле, так как дестабилизация на величину Ей при данной координации одинакова для всех электронов, (экстрастабилизации), равной б = /бД. Наоборот, заполнение электроном eg-орбитали приводит к повышению энергии системы: б = -бД. Зная электронную конфигурацию комплекса, можно вычислить полную энергию стабилизации системы кристаллическим полем (табл. IV. 10 и IV. 11). Данные табл. IV. 10 (стр. 106), сведенные к одинаковому масштабу, позволяют сравнивать между собой энергии стабилизации кристаллическим полем в различных случаях. Эти сравне- ния можно проводить как по вертикали, т. е. с переходом от одного центрального атома к другому для одного и того же типа координации и поля, так и по горизонтали, т. е. для различных типов координации одного и того же центрального атома. Остановимся сначала на сравнении данных табл. IV. 10 по вертикали. Рассмотрим октаэдрическне высокоспиновые комплексы двухвалентных переходных металлов первого большого периода с одинаковыми лигандами (например, водные комплексы). 500\- о о § § я 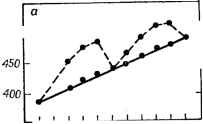 Са SC Ti V Сг МП Fe Со Nt Си Zn 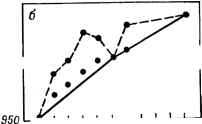 3080,5 - 2095,0 - 1885,5 W76,0 Н66,5 4818,5 1050\- ScTi VCrMnFeCoNiCu7nGa -j 439S,S 3980,5 Рис IX. 1. Двугорбая зависимость теплот образования ки водных комплексов двухвалентных (а) и трехвалентных (б) переходных металлов. Сплошнол линией соединены ДЯ за вычетом энергии экстрастабилизации. Основная часть устойчивости, как уже отмечалось, растет монотонно при переходе от одних металлов к другим слева направо по периодической системе или сверху вниз по табл. IV. 10, т. е. йо мере роста п. Однако энергия дополнительной стабилизации кристаллическим полем при этом изменяется немонотонно: она равна нулю для случаев d°, # и и положительна для промежуточных значений, достигая максимума при d(Cr V+) и c?*(NP+). Следовательно, кривая зависимости устойчивости от атомного номера в этом ряду должна получаться двугорбой с максимумами и минимумами, определяемыми значениями энергии экстрастабилизации (см. табл. IV. 10). Именно такой является представленная на рис. IX. 1 кривая теплот гидратации ДЯ рассматриваемых переходных металлов [2, с. 80]. Там же приведена аналогичная кривая для тех же комплексов переходных металлов в трехвалентном состоянии. Как видим, она имеет лочти такой же двугорбый вид. Если воспользоваться опытными значениями параметра расщепления Д (см. табл. VII. 2) и вычесть из экстремальных значений теплот образования водных комплексов величины энергий экстрастабилизации, определяемых с помощью табл. IV. 10, то получаемые значения ДЯ укладываются на монотонно возрастающую кривую, являющуюся почти прямой (рис. IX. 1). Двугорбая форма кривой теплот образования характерна для широкого класса соединений как в растворах, так и в твердом состоянии с молекулами кристаллизационной воды и без нее. Это хорошее качественное согласие теории с экспериментом сыграло известную роль в развитии теории кристаллического поля в применении к комплексным соединениям. Для тетраэдрических и кубических комплексов зависимость теплот образования от атомного номера должна иметь такой же вид, как и для случая октаэдра, но с некоторыми отличиями в деталях. Для низкоспиновых конфигураций, как легко видеть из табл. IV. 10, аналогичного минимума энергии экстрастабилизации и, следовательно, двугорбой зависимости устойчивости, не существует. Аналогичную закономерность для координационных соединений редкоземельных элементов см. в монографии [94, с. 36]. Сравним также данные по относительным устойчивостям при изменении только спинового состояния, т. е. при переходе от высокоспиновых к низкоспиновым и наоборот. Из табл. IV. 10 видно, что в низкоспиновых конфигурациях энергия эксграстабилизации всегда больше, чём в высокоспиновых. Это и понятно, так как в последнем случае для сохранения параллельного спина электроны помещаются не на стабилизирующих, а на дестабилизирующих уровнях. Однако переход от высокоспиновой к низкоспиновой конфигурации связан также со спариванием электронов. При таком переходе энергия с одной стороны уменьшается за счет экстрастабилизации, а с другой - увеличивается на П при каждом спаривании (см. раздел IV. 3). Например, для низкоспиновой и высокоспиновой конфигурации комплексов Co{d) сравниваться должны значения 0,40Д и 2,40Д - 2П. Член 211 учитывает увеличение энергии за счет спаривания двух электронов. Поэтому низко-спиновая конфигурация при прочих равных условиях является наиболее устойчивой, если Д > П и, наоборот, для Д < П более устойчивой оказывается высокоспиновая конфигурация. Этот -вывод полностью совпадает с полученным ранее (см. разделы IV. 3 и V. 3). Другие данные по использованию понятия энергии экстрастабилизации для исследования свойств координационных систем см. в работах [92, 94, 443] и ссылки в них. IX.2. ВОПРОСЫ РЕАКЦИОННОЙ СПОСОБНОСТИ* Факторы, влияющие на реакционную способность Термином реакционная способность, количественно весьма неопределенным, обычно характеризуют способность системы вступать в ту или иную химическую реакцию с другими системами * Общие вопросы см, также в монографиях [92, 444], 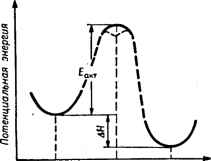 С теоретической точки зрения элементарный акт химической реакции может рассматриваться как переход некоторой системы взаимодействующих частиц из одного стационарного состояния в другое. Оба состояния - начальное и конечное - считаются равновесными, т. е. им соответствуют некоторые минимумы потенциальной энергии системы, являющейся функцией межатомных расстояний. Поэтому при переходе системы из одного состояния в другое потенциальная энергия должна, вообще говоря, сначала повышаться, а затем убывать. Другими словами, между начальным и конечным состояниями системы имеется потенциальный барьер. На рис. IX. 2 схематически * показана зависимость потенциальной энергии от некоторого параметра межатомных расстояний - параметра реакции - вблизи начального состояния А, конечного В и некоторого промежуточного С, соответствующего области наиболее высокой энергии. В состоянии С система может оказаться относительно устойчивой и обладать некоторым дополнительным неглубоким минимумом. Точное квантовомеханическое решение вопроса о ходе химической реакции возможно только в том случае, если известна зависимость поверхности потенциальной энергии от межатомных расстояний вблизи Л и В и во всем промежутке между ними **. Знание этой зависимости позволило бы нам в принципе рассчитать как скорость реакции, так и все ее промежуточные состояния и время жизни каждого из них [314, гл. XVI]. Задача вычисления поверхности потенциальной энергии в таких сложных системах, как неорганические комплексы, в настоящее время практически очень сложна (впрочем, см. [217, 286]). Поэтому остается прибегнуть к упрощенным и полуэмпирическим методам для получения хотя бы некоторых приближенных, качественных и полуколичественных данных о ходе реакции, в частности о строении и энергии начального, конечного и промежуточного А С В Параметр межатомных расстояний Рис. IX. 2. Сечение адиабатического потенциала вблизи начального (А), конечного (J3) и промежуточного (С) состояний химической реакции. Дополнительный пунктирный минимум относится к случаю, когда промежуточное состояние обладает нек1тороЯ относительной устойчивостью. * В действительности потенциальная энергия является многомерной поверхностью, графическое изображение которой на плоскости в общем случае представляется невозможным. ** Предполагается, что элементарный акт реакции проходит как достаточно медленный адиабатический процесс; это, как правило, характерно для реакций в жидкой фазе. В противном случае ее необходимо рассчитывать как квантовомеханическое столкновение [411]. состояний. Такие данные позволили бы определить высоту потенциального барьера- энергию активации реакции (рис. IX. 2) и с помощью некоторого предположения о форме барьера вычислить скорость реакции - один из наиболее важных показателей реакционной способности. Однако в отличие от начального и конечного состояний реакции, которые соответствуют устойчивым химическим объектам и поддаются экспериментальному изучению, промежуточное состояние не всегда поддается экспериментальному исследованию, по крайней мере в интересующей нас области комплексных соединений. Поэтому весьма ценны теоретические представления о строении промежуточного состояния - так называемого активированного комплекса . Такие представления можно получить на основе того или иного предположения о механизме реакции. В больщинстве случаев наблюдаемые реакции с координационными соединениями являются реакциями замещения. Это связано с тем, что обычно реакции протекают в растворах и металлический ион-комплексообразователь окружен (в свободных местах) соль-ватной оболочкой, молекулы которой могут рассматриваться как лиганды. В связи с этим изменения в координационной сфере металла могут считаться процессами замещения одного лиганда другим. В обычной терминологии такое замещение называется нук-леофильным и обозначается Sn- Кроме нуклеофильного, в принципе возможно также электрофильное замещение Se, когда центральный атом замещается другим (см., например, [445]). Впрочем, Лэнгфорд и Грэй предлагают несколько другую классификацию реакций замещения координационных соединений [444, гл. 1]. Различают в основном два механизма реакций нуклеофильного замещения в комплексных соединениях: механизм диссоциации Sjvl и бимолекулярный механизм смещения Sjv2. В первом случае предполагается, что замещаемый лиганд предварительно отрывается от комплекса (комплекс диссоциирует) и затем замещающий занимает его место. Во втором механизме предварительная диссоциация не предполагается; напротив, замещающий и замещаемый лиганды сосуществуют некоторое время совместно в активированном комплексе. При этом замещаемый лиганд под влиянием замещающего сначала смещается со своего равновесного положения, а затем отрывается, освобождая место последнему. Как для одного, так и для другого механизма начальное и конечное состояния системы одинаковы. Существенное различие между этими механизмами выступает лищь при анализе промежуточного состояния. Для реакций 51 последнее является диссоциированным комплексом, у которого не хватает одного лиганда по сравнению с начальным состоянием, а для механизма Sn2 промежуточное состояние представляет собой комплекс, содержащий на один лиганд больше исходного. Для переходных металлов первого большого периода, для которых реализуются комплексы с координационными числами (к. ч.) преимущественно 4, 5 и 6, к. ч. 3 и 7 энергетически менее выгодны, чем 5. Поэтому можно ожидать, что для этих комплексов предпочтительными будут реакции замещения, протекающие по механизму Sjvl для к. ч. 6 и 52 для к. ч. 4. Для переходных металлов следующих периодов, как будет показано ниже, возможность реализации механизма 5jv2 в реакциях замещения увеличивается. Предположив тот или иной механизм реакции, можно приближенно рассчитать энергию промежуточного активированного комплекса и тем самым определить высоту барьера и скорость реакции. Например, по теории кристаллического поля изменение энергии комплекса При переходе в промежуточное состояние складывается из трех частей: 1) изменение энергии притяжения металл - лиганд (уменьшение притяжения и, следовательно, увеличение энергии системы для механизма 5jvl й уменьшение для 5iv2); 2) изменение энергии взаимного отталкивания лигандов (уменьшение для Sfl и увеличение для 5jv2); 3) изменение энергии стабилизации кристаллическим полем. Расчеты можно существенно упростить, если ограничиться учетом только последней части этой энергии - энергии экстрастабилизации (табл. IV. 10 и IV. 11) и полностью игнорировать первые две части. Это упрощение основано на том, что изменения энергий притяжения и отталкивания имеют противоположные знаки и частично компенсируют друг друга. Хотя такое упрощение довольно грубо, оно все же позволяет получить некоторые удовлетворительные качественные результаты без практически сложных расчетов. Таблица IX. 1 Сравнение экспериментальных и теоретических данных по диссоциации некоторых о-фенантролиновых (Phen) и а, а-дипиридиловых (Dipy) комплексов при 25 С [446]
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||