
|
|
|
Главная -> Свойства координационных соединений различных атомов. В свете этого полученный выше результат означает, что корреляция электронов на разных атомах лучше учитывается в Х -методе, чем в методе МО ЛКАО. 101- S 40 Рис. v.9. Сравнение результатов расчета энергии ионизации валентных состояний молекулы SFe по методу ССП-Хц - РВ [236] (в приближении промежуточных состояний) (а) с полученными экспериментально из фотоэлектронных спектров (б), с расчетами по методу ППДП. (в) и в неэмпирическом приближении ССП МО ЛКАО (г). Методом ССП -Х -РВ к настояшему времени проделано большое число расчетов малых, средних и больших молекул (см. обзоры [232, 236]). Из малых и средних молекул упомянем здесь: sol [241. 242]; ClOI [241]; SFg [243]; С,Н, [244]; N СО, NO, CF4, Н2О, СН4. Н2, Liz, CO2, C3O2, N2O, C4H4S, СбНб. NH3, H2O2, P Ps и др. (см. [236]). В качестве примера точности полученных результатов приведем на рис. V. 9 [236, 243} значения энергий ионизации различных валентных состояний молекулы SFe [243] и их сравнение, с одной стороны, с экспериментальными данными по фотоэлектронной 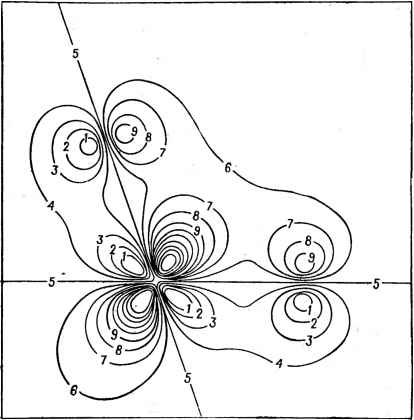 Рис. V. 10. Контурная карта ортонормированиой /е-волновой функции в плоскости О-Мп-О тетраэдрического комплекса МпО, полученной расчетом по методу ССП-Хц-РВ (по работе [236]). Контуры приведены с интервалом в 0,05 от значения -0,2 (контур № 1) до +0,2 (кон- По контуру Jfs 5 проходит узел я-функцви. Расстояние Мп-0 - тур № 9). По контуру Jfs 5 проходит узел Я-функцви. Расстояние равно 3,00 а. е. спектроскопии [245], и, с другой - с неэмпирическими расчетами [246] и с полуэмпирическими расчетами по методу ППДП (с использованием теоремы Купманса). Из рис. V.9 видно, что Х -ме-тод дает наилучшее согласие с экспериментом (метод ППДП дает даже другой порядок следования этих потенциалов). Такие же хорошие результаты получены для всех перечисленных молекул, к тому же со значительно меньшей затратой машинного времени (для расчета молекулы SFe, включая расчет всех промежуточных состояний, на машине ИБМ 360/65 затрачено 6 мин [243], в то время как неэмпирический расчет МО ЛКАО той же молекулы требует от 20 до 30 ч [246]). Аналогично, выявлены преимушества метода при расчете оптических переходов, энергий информации, барьеров внутреннего врашения (в СгНе и в Н2О2), инверсионного барьера в NH3 и др. Еще большие надежды возлагаются на преимущества метода при расчете с его помощью больших молекул, комплексов переходных металлов, биологических макромолекул, молекулярных образований на поверхности, кристаллов и т. д. Подробный расчет проведен для отмеченного выше традиционного объекта проверки методов расчета электронного строения комплексов - MnOi [235]. Всего 15 итераций и 8 мин времени потребовалось для расчета этого комплекса на машине ИБМ 360/65 (а с учетом введенных после этой работы усовершенствований в расчетной процедуре это время сокращается наполовину). При этом полученные результаты, в частности по энергии связи, потенциалам ионизации, энергии электронных переходов и др. находятся в лучшем согласии с экспериментальными данными, чем во всех остальных ме- одах. На рис. V. 10 схематически (в разрезе) иллюстрируется полученная в расчете волновая функция 1е-орбитали, из которой видно образование я-связи типа Mn(3d) - 0(2р). Об этой последней, однако, в методе можно судить лишь косвенно - по симметрии молекулярной орбитали (в отличие от метода МО ЛКАО, в котором благодаря разложению МО по АО образование различных типов связей и степень ковалентности на них видны непосредственно по значениям коэффициентов ЛКАО). Другие комплексы, рассчитанные Х -методом: TiCl, NiF, CuCir, PtCl, Ni(C0)4, Сг(СО)б, Fe(C0)5 и др. (см. [236]). В самое последнее время [247] был рассчитан кластер NiOe с учетом всех 86 электронов и влияния кристаллического окружения (расчет потребовал 30 итерраций и 10 минут времени на машине ИБМ 370/165) с целью объяснения экспериментальных данных по рентгеновской фотоэлектронной спектроскопии кристалла NiO [248]. В результате расчета получено хорошее согласие с этими данными. Расчетные данные [247] позволили решить также важную задачу об адсорбции кислорода на поверхности металла никеля. Из сравнения результатов расчета с фотоэлектронными спектрами адсорбированного кислорода и его концентрационной зависимости [249] удалось установить, что при достаточно больших концентрациях атомы кислорода проникают в кристалл и на поверхности образуются кластеры NiOe [250]. Оценивая метод в целом, можно заключить, что он относится к полуколичественным неэмпирическим методам (в том смысле, что, не считая Ru в нем не используются эмпирические параметры, но гамильтониан системы берется приближенным и решения ап- роксимируются разложением по сферическим областям ). С его помощью можно в хорошем приближении рассчитать электронное строение достаточно больших многоатомных систем. Главное его преимущество перед методом МО ЛКАО -значительно меньшие затраты машинного времени, особенно заметное в случае больших систем. В Х -методе нет проблемы выбора базиса и нет зависимости времени счета от числа функции базиса {n). Поэтому с возрастанием числа атомов время счета растет гораздо медленней, чем в методе МО ЛКАО. К недостаткам метода, которые проявляются непосредственно, следует отнести его неприспособленность к расчету геометрии молекул (Х -метод слабо чувствителен к изменению углов связей) и отсутствие наглядной генеалогической зависимости результатов от строения составлйющих систему атомов, столь важной для широких кругов исследователей. ГЛАВА VI ЭЛЕНТРОННО-НОЛЕБАТЕЛЬНЫЕ (ВИБРОННЫЕ) ВЗАИМОДЕЙСТВИЯ. ЭФФЕКТ ЯНА-ТЕЛЛЕРА Проявления электронно-колебательных {или, короче, виброн-ных) взаимодействий в многоатомных системах, в литературе объединяемые под общим названием эффекта Яна - Теллера, образуют в настоящее время новое быстро развивающееся направление в физике и химии молекул и кристаллов. Рассмотренные выше методы исследования электронного строения координационных соединений основываются преимущественно на предположении, что эти соединения имеют определенную конфигурацию ядер, которую можно считать неизменной при рассмотрении электронного движения. Между тем ядра тоже являются квантовыми микрообъектами, движение которых описывается волновыми функциями, так что их пространственная конфигурация (местонахождение), вообще говоря, определяется лишь вероятностно и зависит от условий измерений. Однако, как будет показано, зачастую возможно разделение электронного и ядерного движений в адиабатическом приближении, при котором электронную структуру рассматривают отдельно от динамики ядер. В тех же случаях, когда такое разделение невозможно, что бывает, главным образом, при наличии электронного вырождения или квазивырождения, ситуация становится очень сложной и приводит к ряду новых эффектов и закономерностей. Изложению некоторых теоретических результатов, достигнутых в этой области, посвящена настоящая глава. Влияние на некоторые спектроскопические и кристаллохимические свойства рассматривается в гл. VH-IX. VI.1. КОЛЕБАТЕЛЬНАЯ ЗАДАЧА В ОТСУТСТВИЕ ЗЛЕКТРОННОГО ВЫРОЖДЕНИЯ Адиабатическое приближение [251; 252, § 129; 253, гл. IV] Для решения задачи о движении ядер введем обозначения: q - для совокупности электронных координат и Q -для ядерных, и представим полный гамильтониан системы в уравнении Шре- дингера . q) = £4(<7. Q) (VI.l) в виде суммы трех частей (VI. 2) где fiq - часть гамильтониана, содержащая только электронные координаты; Яд = - j-Aq - оператор кинетической энергии ядер; F(9,Q)-оператор взаимодействий между электронами и ядрами, включающий члены взаимного отталкивания ядер. Попытаемся решить уравнение (VI. 1) поэтапно. Сначала запишем электронное уравнение [Нд + V {д. Q)] Ф (<?, Q) = е (Q) ф (д. Q) (VI. 3) В этом уравнении координаты ядер Q фигурируют как параметры (а не как переменные интегрирования); для каждого фиксированного значения Q (VI. 3) дает набор решений - собственных значений eft(Q) и собственных функций (f)k(q,Q). Решение полного уравнения (VI. 1) можно искать в виде разложения в ряд по ортонормированиой системе функций 4)(9, Q): (-Z. С)=1;х(<3)фП7. Q) (VI. 4) Коэффициенты этого разложения Xfc(Q). естественно, также зависят от Q. Для их нахождения подставим (VI. 4) в уравнение Шредингера (VI. 1), помножим его слева на ф(<7,Q) и проинтегрируем по всему пространству координат электронов; это дает: Матричный элемент Amh некоторого оператора А ( оператора недиабатичности ) определяется следующим образом: -2 5 ф;;{д, Q)VQif(д, Q)Qjk(Q) rf?] (VI. 6) Таким образом, точное решение уравнения Шредингера для системы может быть задано в виде ряда (VI. 4) с коэффициентами Xm{Q), определяемыми из бесконечной системы зацепляющихся (через члены с Amh) уравнений (VI.5). Решение этой системы встречает большие трудности. Однако в хорошем приближении систему (VI. 5) можно упростить. Рассмотрим вначале случай, когда получаемый при решении электронного уравнения терм eh{Q) не вырожден. Тогда члены mhXhiQ) по (VI. 6) малы по сравнению с остальными членами в уравнениях (VI. 5) и ими можно пренебречь. Действительно, зависимость функций q>k{q,Q) от ядерных координат Q при малых Q можно определить следующим образом. Разложим оператор электронно-колебательного взаимодействия V{q,Q) в ряд по малым Q
|